
İçindekiler
- İdiyopatik Pulmoner Fibrozis Nedir?
- İdiyopatik Pulmoner Fibrozis Sebebi Nedir? Neden Olur?
- IPF Hastalığı Evreleri
- İdiyopatik Pulmoner Fibrozis Risk Faktörleri
- İdiyopatik Pulmoner Fibrozis Tedavisi
- Farmakolojik Tedavi
- Destekleyici Tedavi ve Non-Farmakolojik Yaklaşımlar
- Pulmoner Fibrozis Ne Kadar Yaşar?
- Akciğerde Fibrozis İyileşir Mi?
- Prof. Dr. Levent Alpay’ın Klinik Değerlendirmesi
- Tanıdan İyileşmeye: Süreç Nasıl İlerliyor?
- Sıkça Sorulan Sorular
- Pulmoner fibrozis ne kadar yaşar?
- Akciğerde fibrozis iyileşir mi?
- Sonuç
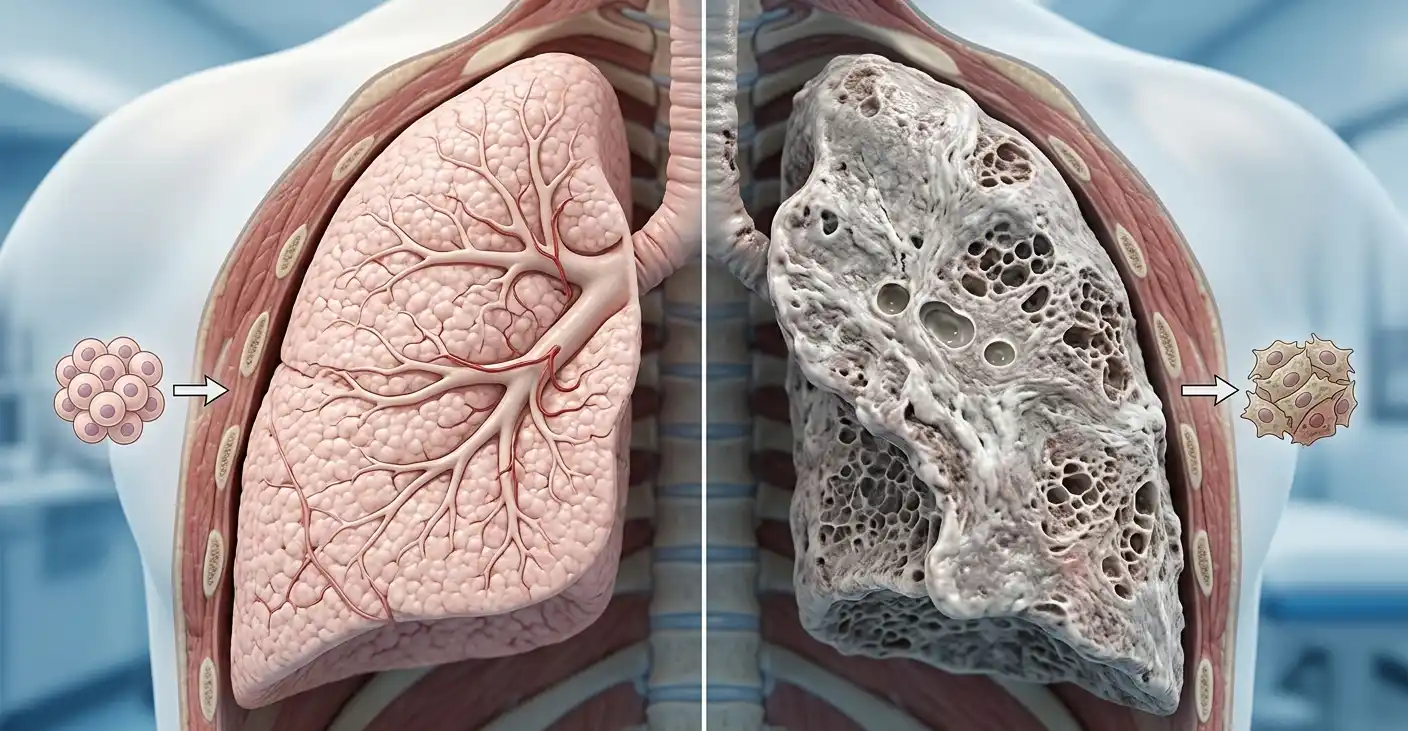
- İdiyopatik pulmoner fibrozis (IPF), akciğerlerdeki hava kesecikleri (alveoller) çevresinde ilerleyici ve geri dönüşümsüz yara dokusu (fibrozis) oluşumuna yol açan, nedeni bilinmeyen kronik bir interstisyel akciğer hastalığıdır.
- Hastalık genellikle 60 yaş üzeri erkeklerde görülür; Am J Respir Crit Care Med’de yayımlanan 2022 ATS/ERS/JRS/ALAT kılavuzuna göre, küresel insidans 100.000 kişide yılda 5.8 vaka, prevalans ise 100.000 kişide 17.7 vakadır.
- Tanı, yüksek çözünürlüklü göğüs BT (HRCT) ve çok disiplinli tartışma (MDD) ile konulur; karakteristik bulgu “usual interstitial pneumonia (UIP)” paternidir.
- Medyan sağkalım tanıdan itibaren 2-3 yıl olup, akut alevlenmeler (AE-IPF) hastalığın en ciddik komplikasyonudur; yıllık insidansı %5-15 arasındadır ve kısa dönem mortalitesi ~%50’dir.
- Antifibrotik ajanlar pirfenidon ve nintedanib, hastalık ilerlemesini yavaşlatır ancak tedavi edici değildir. Son evrede tek etkili seçenek akciğer transplantasyonudur.
İdiyopatik Pulmoner Fibrozis Nedir?
İdiyopatik pulmoner fibrozis, akciğer parankiminde ilerleyici, geri dönüşümsüz fibrotik değişikliklerle karakterize, nedeni bilinmeyen (idiopatik) bir kronik interstisyel akciğer hastalığıdır. Hastalığın temel patofizyolojisi, tekrarlayan alveoler epitel hasarı sonrası anormal yara iyileşme yanıtıdır.
Bu süreçte fibroblastlar aktivasyona uğrar, myofibroblastlara farklılaşır ve aşırı miktarda ekstraselüler matris (özellikle kollagen) birikimine yol açar. Sonuçta alveoler yapı bozulur, gaz değişimi bozukluğu gelişir ve akciğer fonksiyonları geri dönüşümsüz şekilde azalır.
Prof. Dr. Levent Alpay’a göre, IPF tanısı alan bir hastanın en kritik ihtiyacı, hastalığın doğal seyrini ve mevcut tedavi seçeneklerinin sınırlarını gerçekçi şekilde anlamasıdır; çünkü erken ve doğru bilgilendirme, yaşam kalitesi yönetimini doğrudan etkiler.
İdiyopatik Pulmoner Fibrozis Sebebi Nedir? Neden Olur?
İdiyopatik pulmoner fibrozis için tek bir kesin neden belirlenememiştir. “İdiyopatik” tanımlaması, hastalığın nedeni bilinmediğini ifade eder.
Ancak son on yılda yapılan araştırmalar, hastalığın multifaktöriyel bir etiyolojiye sahip olduğunu göstermektedir. Front Pharmacol’de 2022 yılında yayımlanan güncel bir derlemede, IPF’nin tekrarlayan alveoler epitel hasarı ve sonrasında anormal yara iyileşme yanıtı sonucu geliştiği vurgulanmaktadır. Bu anormal yanıtta, TGF-β gibi büyüme faktörleri, Wnt/β-katenin sinyal yolağı ve epitel-mezenkimal geçiş (EMT) kritik roller oynar.
Hastalığın gelişiminde şu faktörlerin bir araya geldiği düşünülmektedir:
● Genetik yatkınlık: MUC5B promotör varyantı (rs35705950), TERT ve TERC gen mutasyonları ailevi ve sporadik IPF ile ilişkilidir. Yaklaşık %15-20 olguda aile öyküsü mevcuttur.
● Çevresel ve mesleksel maruziyetler: Metal tozları (bakır, kurşun), ahşap tozları, silika, asbest ve hayvan/bitkisel tozları riski artırabilir.
● Mikrobiyal faktörler: Epstein-Barr virüsü, hepatit C virüsü ve herpes virüsleri ile olası ilişki incelenmektedir ancak nedensellik henüz kanıtlanmamıştır.
● Gastroözofageal reflü (GERD): Mikroaspirasyonun alveoler epitel hasarına katkıda bulunduğu hipotezi mevcuttur. 2022 ATS/ERS/JRS/ALAT kılavuzunda, antiasit tedavisi ve antireflü cerrahisi için IPF’de solunum sonuçlarını iyileştirme amacıyla koşullu öneri karşıtı (conditional recommendation against) yapılmıştır.
● Sigara: Am J Respir Crit Care Med’de 1997 yılında yayımlanan çok merkezli bir vaka-kontrol çalışmasında, sigara içmiş olanlarda IPF riski hiç içmemişlere göre anlamlı şekilde yüksek bulunmuştur (OR: 1.6; %95 GA: 1.1-2.4); eski sigara içicilerinde risk daha da belirgindir (OR: 1.9; %95 GA: 1.3-2.9).
Prof. Dr. Levent Alpay, klinik pratiğinde IPF tanısı alan hastaların büyük çoğunluğunun 60 yaş üzeri, erkek ve sigara öyküsü olan bireylerden oluştuğunu gözlemliyor. Bu demografik örüntü, hastalığın hem çevresel hem de genetik faktörlerin uzun yıllar içinde birikmesiyle ortaya çıktığı teorisini desteklemektedir.
IPF Hastalığı Evreleri
İdiyopatik pulmoner fibrozis evrelemesi, hastalığın prognozunu belirlemek ve tedavi stratejisini şekillendirmek için kullanılır. En yaygın kullanılan sistem GAP (Gender-Age-Physiology) İndeksi ve evreleme sistemidir. Ann Intern Med’de 2012 yılında yayımlanan ve üç farklı merkezde (California, Minnesota, İtalya) doğrulanan bu sistem, dört değişkeni kullanır: cinsiyet (G), yaş (A), zorlu vital kapasite (FVC) ve karbonmonoksit difüzyon kapasitesi (DLco) (P).
| GAP Evresi | Özellikler | 1 Yıllık Mortalite | 3 Yıllık Mortalite |
| Evre I | Yaş ≤60, FVC ≥%75, DLco ≥%55 (erkeklerde daha iyi prognoz) | ~%6 | ~%20-25 |
| Evre II | Orta düzeyde bozulmuş parametreler | ~%16 | ~%35-45 |
| Evre III | Yaş >70, FVC <%50, DLco <%35 | ~%39 | ~%55-65 |
Chest dergisinde 2016 yılında yayımlanan bir çalışmada, GAP evresi ölüm veya akciğer transplantasyonunu öngörmede başarılı olmakla birlikte, 6 ayda FVC veya DLco’da ≥%10’luk düşüş olmasının, evre dikkate alındıktan sonra bile bağımsız olarak kötü prognozu öngördüğü gösterilmiştir (FVC için HR: 1.37; DLco için HR: 1.30). Bu bulgu, statik evrelemenin yanında dinamik fonksiyon takibinin de hayati önem taşıdığını ortaya koymaktadır.
Prof. Dr. Levent Alpay’a göre, GAP evrelemesi klinik pratiğinde hızlı ve uygulanabilir bir araçtır; ancak hastanın egzersiz kapasitesi, 6 dakika yürüme testi sonuçları ve akut alevlenme öyküsü de prognostik değerlendirmede mutlaka dikkate alınmalıdır.

İdiyopatik Pulmoner Fibrozis Risk Faktörleri
İdiyopatik pulmoner fibrozis gelişiminde bilinen ve şüphelenilen risk faktörleri şunlardır:
● Yaş ve cinsiyet: 60 yaş üzeri erkeklerde görülme sıklığı belirgin şekilde daha yüksektir. BMC Pulm Med’de 2023 yılında yayımlanan Kore’deki bir kohort çalışmasında, ortalama tanı yaşı 65.9 yıl ve erkeklerin oranı %67.5 olarak bildirilmiştir.
● Sigara içme öyküsü: Bağımsız ve güçlü bir risk faktörüdür. 21-40 paket-yıl maruziyetinde risk anlamlı şekilde artar (OR: 2.3; %95 GA: 1.3-3.8).
● Aile öyküsü: Ailevi interstisyel akciğer hastalığı (FIP) olarak adlandırılan form, genetik mutasyonlarla ilişkilidir. MUC5B rs35705950 varyantı, riski 6-20 kat artırabilir.
● Mesleksel ve çevresel maruziyetler: Metal işleme, tarım, hayvancılık, marangozluk ve madencilik gibi mesleklerde çalışanlarda IPF riski artışı gözlemlenmiştir.
● Virüs enfeksiyonları: EBV, CMV, HHV-7 ve hepatit C virüsü ile olası ilişki araştırılmaktadır.
● Gastroözofageal reflü: Mikroaspirasyonun alveoler hasara yol açtığı teorisi, ancak 2022 kılavuzunda antireflü tedavisi için IPF’de öneri yapılmamaktadır.
● Diabetes mellitus: Bazı epidemiyolojik çalışmalarda IPF ile tip 2 diyabet arasında pozitif ilişki bildirilmiştir.
İdiyopatik Pulmoner Fibrozis Tedavisi
İdiyopatik pulmoner fibrozis tedavisi, hastalık ilerlemesini yavaşlatmaya, semptomları kontrol etmeye ve yaşam kalitesini korumaya odaklanır. Maalesef günümüzde hastalığı tamamen tedavi eden bir yöntem bulunmamaktadır.
Farmakolojik Tedavi
2022 ATS/ERS/JRS/ALAT kılavuzuna göre, IPF’de güçlü öneri (strong recommendation) ile önerilen tek ilaçlar pirfenidon ve nintedanib’tir. Her ikisi de antifibrotik etki mekanizmasıyla hastalık ilerlemesini yavaşlatır:
| Özellik | Pirfenidon | Nintedanib |
| Mekanizma | TGF-β ve TNF-α inhibisyonu, antioksidan etki | Tirozin kinaz inhibitörü (VEGF, FGF, PDGF reseptörleri) |
| Etkinlik | FVC düşüşünü yavaşlatır, %10 FVC düşüş riskini azaltır | FVC düşüşünü yavaşlatır, akut alevlenme riskini azaltır |
| Yaygın yan etkiler | Bulantı, ishal, fotosensitivite, cilt döküntüsü | İshal, bulantı, karın ağrısı, karaciğer enzim yüksekliği |
| Bırakma oranı | ~%16-17 ( advers etki nedeniyle) | ~%16-20 (advers etki nedeniyle) |
BMC Pulm Med’de 2021 yılında yayımlanan bir meta-analizde, antifibrotik tedavinin (pirfenidon ve nintedanib birlikte) FVC düşüşünde standartlaştırılmış etki büyüklüğü -0.305 (p < 0.001) olarak bulunmuş ve tüm nedenlere bağlı mortalitede risk oranı 0.701 (p = 0.008) lehine anlamlı azalma gösterilmiştir. Chest’te 2024 yılında yayımlanan CleanUP-IPF çalışmasının post-hoc analizinde ise, nintedanib kullanan hastalarda 12 aylık FVC düşüşünün pirfenidona göre daha yavaş olduğu (ortalama fark: 106 mL; %95 GA: 34-178) ancak genel sağkalım ve hastaneye yatış açısından anlamlı fark olmadığı bildirilmiştir.
Prof. Dr. Levent Alpay’a göre, pirfenidon ve nintedanib arasındaki seçim, hastanın komorbiditelerine, yan etki toleransına ve ilaç etkileşimlerine göre bireyselleştirilmelidir. Örneğin, koroner arter hastalığı öyküsü olan hastalarda nintedanib kullanımı dikkatli değerlendirilmelidir.
Destekleyici Tedavi ve Non-Farmakolojik Yaklaşımlar
● Oksijen tedavisi: Dinlenme veya egzersiz sırasında hipoksemi gelişen hastalarda uzun dönem oksijen tedavisi önerilir.
● Pulmoner rehabilitasyon: Egzersiz kapasitesini, dispne şiddetini ve yaşam kalitesini iyileştirir.
● Akut alevlenme yönetimi: Yüksek doz kortikosteroidler ve antibiyotikler yaygın olarak kullanılır ancak net kanıta dayalı veri yoktur; kısa dönem mortalite ~%50’dir.
● Akciğer transplantasyonu: Son evre IPF’de tek etkili tedavidir. Ann Thorac Surg’de 2007 yılında yayımlanan bir çalışmada, IPF için transplantasyon sonrası sağkalım oranları 1 yılda %73, 3 yılda %56 ve 5 yılda %44 olarak bildirilmiştir. J Heart Lung Transplant’te 2016 yılında yayımlanan bir analizde, çift akciğer transplantasyonu ile tek akciğer transplantasyonu arasında anlamlı sağkalım farkı bulunmamıştır.
Pulmoner Fibrozis Ne Kadar Yaşar?
İdiyopatik pulmoner fibrozis prognozu, tanı anındaki evre, fonksiyon durumu ve akut alevlenme gelişimine bağlı olarak değişkenlik gösterir.
Am J Respir Crit Care Med’de 2011 yılında yayımlanan bir derlemede, IPF medyan sağkalımının 2-3 yıl olduğu vurgulanmaktadır. Ancak bu ortalama değer, bireysel farklılıkları gizleyebilir; bazı hastalar tanıdan sonra 5-10 yıl yaşarken, bazılarında hastalık çok daha hızlı ilerleyebilir.
Thorax dergisinde 1998 yılında yayımlanan, New Mexico Interstisyel Akciğer Hastalığı Kayıt Defteri’nden elde edilen verilere göre, genel popülasyondaki IPF hastalarında medyan sağkalım 4.2 yıl olarak bulunmuştur. Bu fark, hastane bazlı referans merkezlerindeki hastaların genel popülasyondakilerden daha genç ve daha az komorbid olmasıyla açıklanabilir.
GAP evrelemesine göre sağkalım beklentisi şöyledir:
● GAP Evre I: 1 yıllık mortalite ~%6, 3 yıllık mortalite ~%20-25
● GAP Evre II: 1 yıllık mortalite ~%16, 3 yıllık mortalite ~%35-45
● GAP Evre III: 1 yıllık mortalite ~%39, 3 yıllık mortalite ~%55-65
Eur Respir J’de 2011 yılında yayımlanan bir çalışmada, akut alevlenme geçiren IPF hastalarında yatış sırasında mortalite oranı %50 olarak bildirilmiş ve akut alevlenmenin tanıdan sonraki 1 ve 5 yıllık sağkalım oranlarını sırasıyla %56.2 ve %18.4’e düşürdüğü gösterilmiştir. Sci Rep’te 2024 yılında yayımlanan bir meta-analizde, 1 yıllık akut alevlenme insidansı %9, 2 yıllık %13 ve 3 yıllık %19 olarak hesaplanmıştır.
Prof. Dr. Levent Alpay, klinik pratiğinde prognozu değerlendirirken yalnızca GAP evresine değil, aynı zamanda hastanın 6 dakika yürüme testi sonuçlarına, oksijen satürasyon eğilimlerine ve akut alevlenme öyküsüne baktığını vurguluyor. Bu bütüncül yaklaşım, hastaya ve ailesine daha gerçekçi bir beklenti çerçevesi sunar.
Akciğerde Fibrozis İyileşir Mi?
Akciğerde fibrozis, günümüzde bilinen tıbbi yöntemlerle iyileştirilemez ve geri döndürülemez. Am J Physiol’de 1990 yılında yayımlanan temel bir derlemede, akciğer fibrozisinin genellikle geri dönüşümsüz (irreversible) olduğu, ancak fibroblastların “aktive” durumunun eksüdat temizlendikten ve reepitelizasyon sağlandıktan sonra tersine çevrilebileceği belirtilmektedir. Ancak IPF’de, otonom fibroproliferatif yanıt ve devam eden hasar nedeniyle bu onarım mekanizması bozulur.
Front Pharmacol’de 2022 yılında yayımlanan güncel bir derlemede, IPF’nin patogenezi şöyle özetlenmektedir: “Kalıcı myofibroblast fenotipi, aşırı ekstraselüler matris birikimine ve anormal akciğer onarımına yol açar; bu durum doku skar oluşumu, alveoler yapı bozulması ve geri dönüşümsüz akciğer fonksiyon kaybına neden olur.” Pirfenidon ve nintedanib ile yapılan tedaviler, akciğer fonksiyon düşüşünü azaltma ve hastalık ilerlemesini yavaşlatma açısından anlamlı olmakla birlikte, hastalığı tedavi etmemektedir.
Bu nedenle, erken tanı ve erken antifibrotik tedavi başlanması kritik öneme sahiptir. Tedavinin amacı, mevcut fonksiyonu korumak ve ilerlemeyi mümkün olduğunca yavaşlatmaktır. Son evrede tek etkili seçenek, akciğer transplantasyonudur.
Prof. Dr. Levent Alpay’ın Klinik Değerlendirmesi
“Klinik pratiğimde sıklıkla şunu gözlemliyorum: İdiyopatik pulmoner fibrozis tanısı alan hastaların çoğu, tanı konulduğunda hastalığın ne kadar ciddi olduğunu tam olarak kavrayamıyor ve internette karşılaştıkları ‘mucize tedavi’ vaatlerine yönelme eğiliminde olabiliyor. Prof. Dr. Levent Alpay’a göre, bu hastalar için en kritik adım, tanı anında gerçekçi ve şeffaf bir bilgilendirme yapılmasıdır. Hastalığın ilerleyici ve geri dönüşümsüz doğasını, antifibrotik ilaçların sınırlarını ve akciğer transplantasyonunun ne zaman gündeme gelmesi gerektiğini açıkça konuşmak, hem tedavi uyumunu artırır hem de hastanın yaşam kalitesi yönetimini olumlu yönde etkiler. Erken evrede pirfenidon veya nintedanib başlanması, fonksiyon kaybını yavaşlatma açısından her geçen ay değerli zaman kaybettirir.”
Tanıdan İyileşmeye: Süreç Nasıl İlerliyor?
İdiyopatik pulmoner fibrozis süreci, ilk şikayetlerden tanıya ve tedaviye kadar şu aşamalardan oluşur:
- İlk Muayene ve Değerlendirme: Hastada aşırı yorgunluk, eforla artan nefes darlığı (dispne), kuru öksürük ve parmak uçlarında şişlik (clubbing) varsa IPF şüphesi gündeme gelir. Sigara öyküsü, mesleksel maruziyetler ve aile öyküsü detaylı şekilde sorgulanır. Prof. Dr. Levent Alpay, bu aşamada hastanın beklentilerini ve hastalık hakkındaki ön bilgilerini anlamanın, sonraki tedavi uyumunu doğrudan etkilediğini vurguluyor.
- Tetkikler ve Görüntüleme: Yüksek çözünürlüklü bilgisayarlı tomografi (HRCT) tanı için hayati öneme sahiptir. Karakteristik “usual interstitial pneumonia (UIP)” paterni: periferal, bazal tutulum, retiküler opasiteler, traksiyon bronşiektaziler ve bal peteği (honeycombing) değişikliklerini içerir. Spirometride restriktif patern (FVC ve FEV1 düşüklüğü, normal ve artmış FEV1/FVC oranı), gaz difüzyon testinde DLco düşüklüğü görülür. 2022 ATS/ERS/JRS/ALAT kılavuzunda, uygun klinik bağlamda HRCT’de probable UIP paterni görülmesi, doku onayına gerek kalmadan tanıyı destekleyebilir.
- Tedavi Süreci: Tanı ve GAP evrelemesi sonrası, hastaya antifibrotik tedavi (pirfenidon veya nintedanib) başlanır. İlaç seçimi, yan etki profili, komorbiditeler ve hasta tercihine göre kişiselleştirilir. Aynı anda oksijen tedavisi, pulmoner rehabilitasyon ve reflü yönetimi gibi destekleyici önlemler planlanır.
- Hastanede Geçirilen Süre: IPF tanısı ve tedavi başlangıcı genellikle ayaktan takiple yürütülür. Akut alevlenme veya solunum yetmezliği gelişen hastalar hastaneye yatırılarak yüksek doz kortikosteroid, oksijen ve gerekirse non-invaziv ventilasyon desteği alır.
- Taburcu Sonrası İyileşme ve Takip: Hastalar düzenli aralıklarla (genellikle 3-6 ayda bir) poliklinik kontrole çağrılır. Her ziyarette FVC, DLco, 6 dakika yürüme testi ve oksijen satürasyonu ölçülür. Fonksiyonlarda ≥%10’luk düşüş, tedavi değişikliği veya transplantasyon değerlendirmesi için işaret olabilir. Prof. Dr. Levent Alpay’a göre, bu düzenli takip, hastalık ilerlemesinin erken fark edilmesi ve zamanında müdahale açısından kritiktir.
Sıkça Sorulan Sorular
Pulmoner fibrozis ne kadar yaşar?
Medyan sağkalım tanıdan itibaren 2-3 yıldır ancak bireysel farklılıklar büyüktür. GAP Evre I hastalarında 1 yıllık sağkalım oranı ~%94, Evre III hastalarında ~%61’dir. Akut alevlenme gelişimi prognozu ciddi şekilde kötüleştirir.
Akciğerde fibrozis iyileşir mi?
Günümüzde akciğerde fibrozis tam olarak iyileştirilemez. Antifibrotik ilaçlar ilerlemeyi yavaşlatır ancak mevcut hasarı geri döndürmez. Erken tanı ve tedavi, fonksiyon kaybını mümkün olduğunca yavaşlatmak için kritiktir.
Sonuç
İdiyopatik pulmoner fibrozis, akciğerlerde ilerleyici ve geri dönüşümsüz fibrotik değişikliklerle seyreden, nedeni bilinmeyen ciddi bir interstisyel akciğer hastalığıdır. Tanı HRCT ve çok disiplinli değerlendirme ile konulur; GAP evrelemesi prognozu belirlemede kullanılır. Antifibrotik ajanlar pirfenidon ve nintedanib hastalık ilerlemesini yavaşlatır ancak tedavi edici değildir. Akut alevlenmeler hastalığın en ciddik komplikasyonudur. Son evrede akciğer transplantasyonu tek etkili seçenektir. Prof. Dr. Levent Alpay’a göre, erken tanı, gerçekçi bilgilendirme ve düzenli takip, bu hastalıkta yaşam kalitesi ve süresini etkileyen en önemli faktörlerdir.
İdiyopatik pulmoner fibrozis hakkında kişisel durumunuzu değerlendirmek ve sorularınızı yanıtlamak için Prof. Dr. Levent Alpay ile görüşebilirsiniz. Medistate Çekmeköy ve Kavacık Hastanesi’ndeki muayenehane randevusu için +90 532 331 04 78 numaralı hattı arayabilirsiniz.
Kaynaklar
- Raghu G. et al., Am J Respir Crit Care Med, 2022 — Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis: ATS/ERS/JRS/ALAT Clinical Practice Guideline
- Fan Y. et al., Ann Am Thorac Soc, 2026 — Incidence Rate and Prevalence of Idiopathic Pulmonary Fibrosis in the United States 2017-2022
- Ley B. et al., Am J Respir Crit Care Med, 2011 — Clinical Course and Prediction of Survival in Idiopathic Pulmonary Fibrosis
- Ley B. et al., Ann Intern Med, 2012 — A Multidimensional Index and Staging System for Idiopathic Pulmonary Fibrosis (GAP Index)
- Mei Q. et al., Front Pharmacol, 2022 — Idiopathic Pulmonary Fibrosis: An Update on Pathogenesis
- Baumgartner KB. et al., Am J Respir Crit Care Med, 1997 — Cigarette Smoking: A Risk Factor for Idiopathic Pulmonary Fibrosis
- Finnerty JP. et al., BMC Pulm Med, 2021 — Efficacy of Antifibrotic Drugs in IPF: Systematic Review and Meta-Analysis
- Kim JS. et al., Chest, 2024 — Comparison of Pirfenidone and Nintedanib: Post Hoc Analysis of CleanUP-IPF
- Song JW. et al., Eur Respir J, 2011 — Acute Exacerbation of IPF: Incidence, Risk Factors and Outcome
- Wang Y. et al., Sci Rep, 2024 — Incidence of Acute Exacerbation of IPF: Systematic Review and Meta-Analysis
- Mason DP. et al., Ann Thorac Surg, 2007 — Lung Transplantation for Idiopathic Pulmonary Fibrosis
- Chauhan D. et al., J Heart Lung Transplant, 2016 — Post-Transplant Survival in IPF: Single vs. Double Lung Transplantation
- Kistler KD. et al., BMC Med, 2014 — Lung Transplantation in Idiopathic Pulmonary Fibrosis: Systematic Review
- Lee JH. et al., BMC Pulm Med, 2023 — Epidemiology and Comorbidities in IPF: Nationwide Cohort Study (South Korea)
- Salisbury ML. et al., Chest, 2016 — GAP Index Stage for Predicting Future Lung Function Decline
- Crouch E., Am J Physiol, 1990 — Pathobiology of Pulmonary Fibrosis
- Ryerson CJ. et al., Eur Respir J, 2015 — Acute Exacerbation of IPF: Shifting the Paradigm
- Richeldi L., Ann Am Thorac Soc, 2015 — Time for Prevention of Idiopathic Pulmonary Fibrosis Exacerbation
